
-
Product Name
SMN antibody
- Documents
-
Description
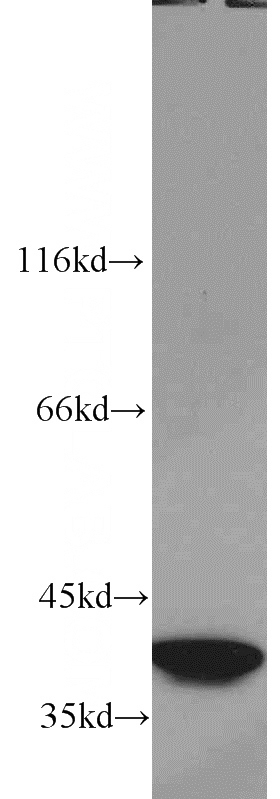
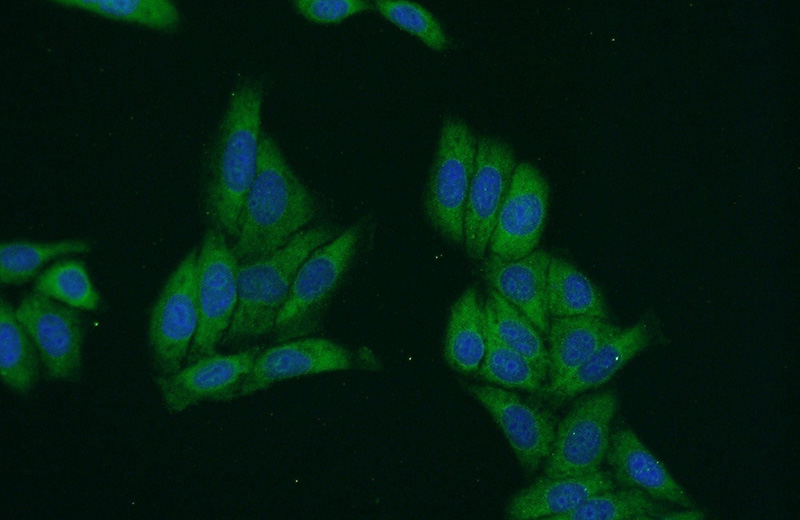
SMN Rabbit Polyclonal antibody. Positive IF detected in HepG2 cells. Positive WB detected in K-562 cells, HEK-293 cells, HepG2 cells, Jurkat cells. Observed molecular weight by Western-blot: 38 kDa
-
Tested applications
ELISA, WB, IF
-
Species reactivity
Human; other species not tested.
-
Alternative names
C BCD541 antibody; Component of gems 1 antibody; FLJ76644 antibody; Gemin 1 antibody; SMN antibody; SMN1 antibody; SMN1 antibody; SMN antibody; SMN2 antibody; SMNC antibody; SMNT antibody; Survival motor neuron protein antibody
-
Isotype
Rabbit IgG
-
Preparation
This antibody was obtained by immunization of SMN recombinant protein (Accession Number: NM_022875). Purification method: Antigen Affinity purified.
-
Clonality
Polyclonal
-
Formulation
PBS with 0.02% sodium azide and 50% glycerol pH 7.3.
-
Storage instructions
Store at -20℃. DO NOT ALIQUOT
-
Applications
Recommended Dilution:
WB: 1:1000-1:10000
IF: 1:20-1:200
-
Validations
K-562 cells were subjected to SDS PAGE followed by western blot with Catalog No:115394(SMN2 antibody) at dilution of 1:2000
Immunofluorescent analysis of HepG2 cells using Catalog No:115394(SMN2 Antibody) at dilution of 1:50 and Alexa Fluor 488-congugated AffiniPure Goat Anti-Rabbit IgG(H+L)
-
Background
Spinal muscular atrophy (SMA) is an autosomal recessive neurodegenerative disease characterized by loss of anterior horn cells in the spinal cord and concomitant symmetrical muscle weakness and atrophy (PMID: 16364894 ). SMA is caused by deletion or mutations of the survival motor neuron (SMN1) gene. SMA patients lack a functional SMN1 gene, but they possess an intact SMN2 gene, which though nearly identical to SMN1, is only partially functional (PMID: 17355180). A large majority of SMN2 transcripts lack exon 7, resulting in production of a truncated, less stable SMN protein (PMID: 10369862). The level of SMN protein correlates with phenotypic severity of SMA.
Related Products / Services
Please note: All products are "FOR RESEARCH USE ONLY AND ARE NOT INTENDED FOR DIAGNOSTIC OR THERAPEUTIC USE"