
Overview
NovoPro provides a successful guaranteed, one stop "Gene-to-Structure“ crystallography service. From gene synthesis, protein production, crystallization to diffraction screening as well as structure analysis. We are trying to give your biology project every chance of success, with utmost convenient.
Structure 1K project

The NovoPro Structure 1K project is aiming to solve 1000 structures in the next 5 years for. Any protein is welcome to inquiry and join our project, enjoy a special package price and fully risk-free policy.
| Gene-to-Structure Complete package | 9,998-15,000 USD |
|---|---|
| Timeline | 2-6 months |
| Standards |
FREE-If-Fail Guarantee Meet Protein Date Bank (PDB) requirements Rfree <0.3, Rwork <0.25 |
Advantages
No pre-payment, free-if-fail policy
Top-tier team, overall success rate>60%
Clients hold full intellectual property rights
One-stop and fast service, up to 2 months.
High quality data gathered from SSRF*, up to 1.0 A resolution
*SSRF: The Shanghai Synchrotron Radiation Facility, which operates at 3.5 GeV, the highest energy of any synchrotron other than the Big Three facilities: SPring-8 in Hyōgo Prefecture, Japan (8 GeV), ESRF in Grenoble, France (6 GeV) and APS at Argonne National labs, United States (7 GeV).

The Shanghai Synchrotron Radiation Facility
Workflow
For an accurate and efficient communication, please download Structure Request Form and email it back to us. All the target protein will be kept as confidential information. A confidentiality agreement/NDA can also be signed prior to the request form, please contact us if needed.
1. Download and fill Structural Biology Form, send it back to the specified email
2. A project proposal and quote will be provided within 48h from submission of the information form
3. Project agreement reached and the project starts
| Procedures | Timeline | Note | |
| 1 | Gene Synthesis | 1-2 Weeks | Free construct design and condon optimization |
| 2 | Protein Expression and Purification | 2-4 Weeks | Activity test supported |
| 3 | Crystallization | 2-8 Weeks | Wizard robotics screening, 1500+ conditions |
| 4 | Diffraction Data Collection | 2-8 Weeks | Most advanced Synchrotron facility, SSRF |
| 5 | Phasing, Refinement and Analysis | 2-3 Weeks | Molecular Replacement, Multiple/Single Isomorphous Replacement, or Multiple/Single Anomalous Diffraction |
| 6 | PDB Submission and Informatics Analysis | 1-2 weeks | In-depth analysis of crystal structures on your requirements |
Successful cases
2016.10.10 Signed the project agreement
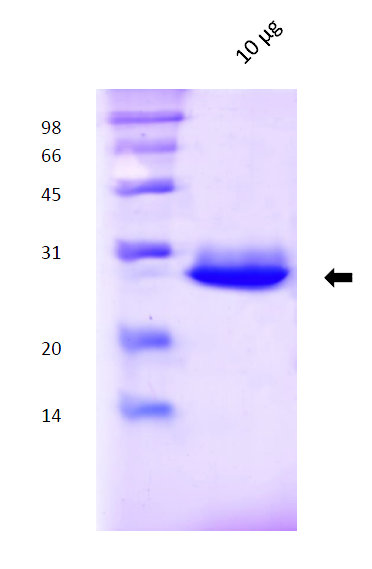
2016.11.13 Successfully prepared the protein (Purity>95%, denature-refolding)
2016.11.27 Crystal screening and optimization
2016.12.29 Data collection at SSRF
2017.01.04 Structure determination finished, Resolution at 1.7Å, Rwork=0.13, Rfree=0.162
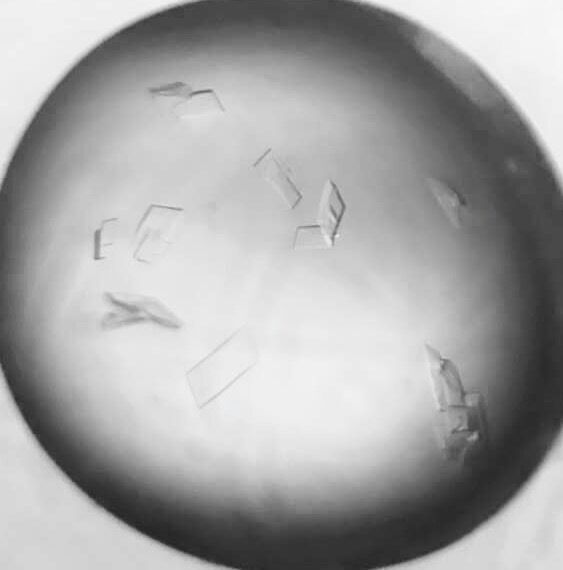

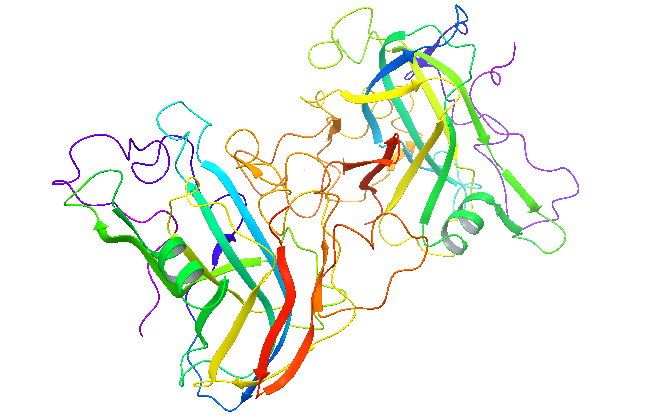
More and more structures are solved at NovoPro…
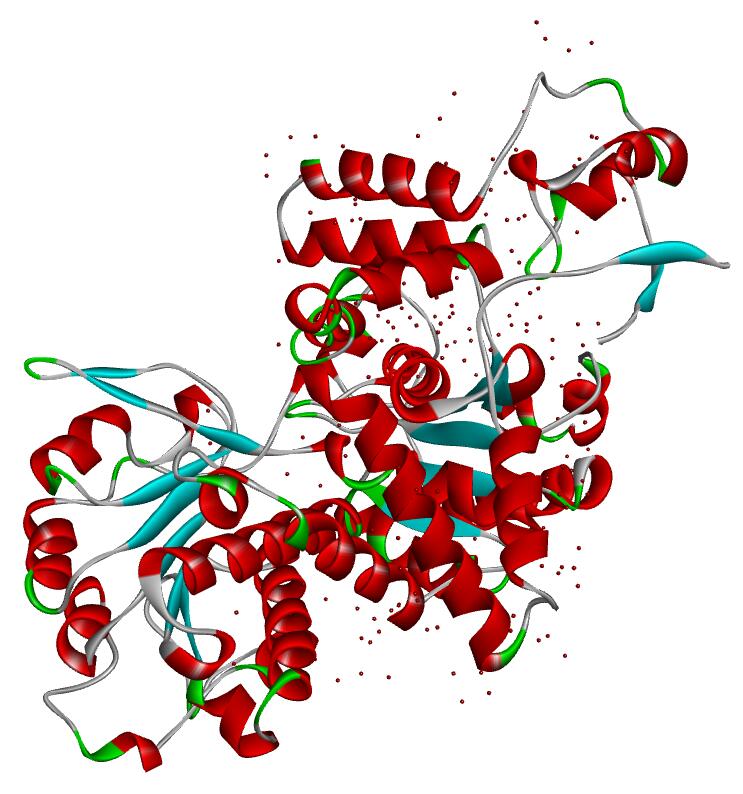
Resolution: 2.0Å; space group: P43212
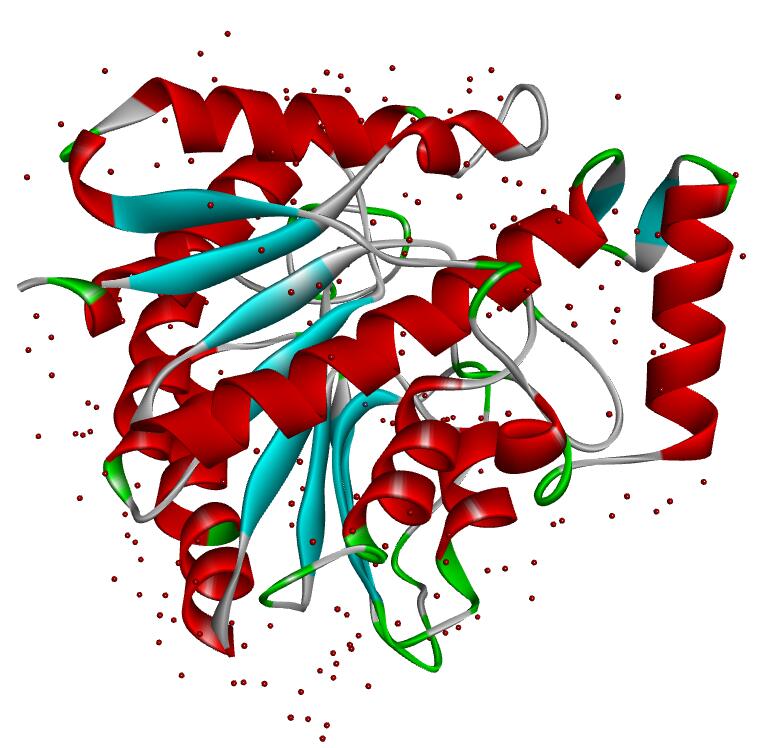
Resolution: 2.19Å; space group: P41
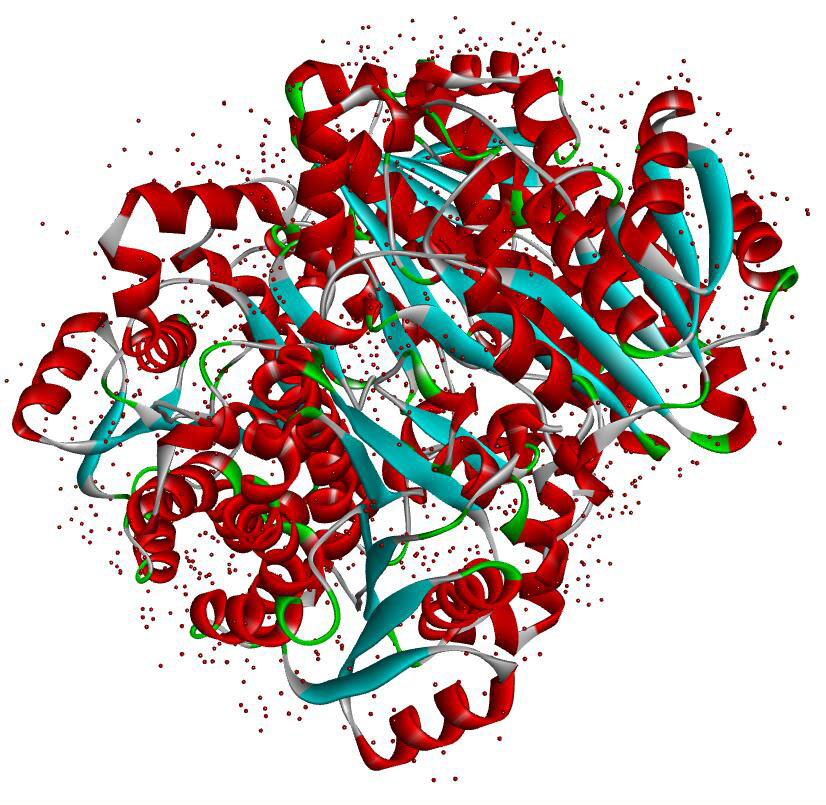
Resolution: 1.8Å; space group: P1
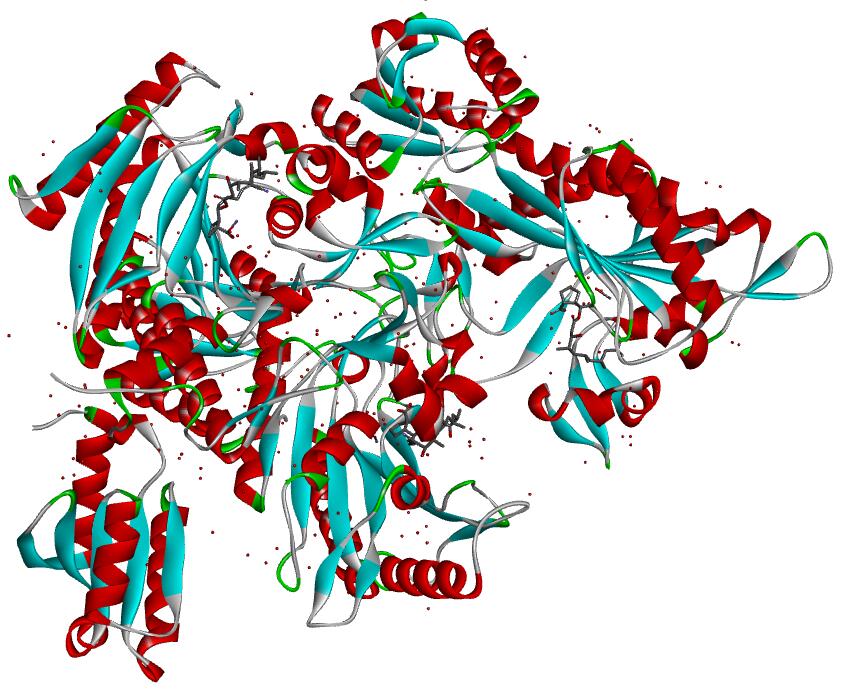
Resolution: 2.0Å; compound with a small molecular
Deliverables
1. Plasmid for recombinant protein production (optional).
2. Purified recombinant protein for activity test (optional).
3. Project report of experimental details
4. Protein structure and informatics analysis results
Detailed project reports
1. Recombinant protein
- 1.1 Plasmid:vector map, cloning strategy, sequencing data;
- 1.2 Protein expression: bacterial strain; expression conditions: temperature, induction time, induction concentration;
- 1.3 Protein purification: methods and results (gel filtration and SDS-PAGE images);
2. Crystallization
- 2.1 Protein Crystallization: protein concentration, buff, methods, temperature and other conditions
- 2.2 Diffraction Data collection:cryo-protectant; X-ray; wavelength; data processing software
- 2.3 Phasing:methods and software
- 2.4 Refinement and Analysis:methods and software
3. Structure
- 3.1 Protein structure .pdb files
- 3.2 wwPDB X-ray structure validation report
- 3.3 .sca, .log, .mtz files generated during data analysis
Data collection and refinement statistics will be provided as followings:
| Data collection | |
| Wavelength (Å) | |
| Space group | |
| Cell dimensions | |
| a, b, c (Å) | |
| α, β, γ (°) | |
| Resolution (Å) | |
| Rmeas (%) | |
| Rmrgd-F (%) | |
| I / σI | |
| Completeness (%) | |
| Multiplicity | |
| Reflections | |
| Unique reflections | |
| Refinement | |
| Resolution (Å) | |
| No. reflections | |
| Rwork / Rfree | |
| No. atoms | |
| Protein | |
| Ligand/ion | |
| Water | |
| B-factors | |
| Protein | |
| Ligand/ion | |
| Water | |
| Ramachandran | |
| Favored (%) | |
| Allowed (%) | |
| Outlier (%) | |
| R.m.s. deviations | |
| Bond lengths (Å) | |
| Bond angles (°) |
Frequently Asked Questions(FAQs)
- 1. Who do I contact for questions?
-
Answer
For enquiries about any requests that have been submitted recently please contact support@novoprolabs.com.
For enquiries about crystal data you have already received you may also contact the person who was concerned with this particular structure.
- 2. How much should I pay if the project fails or does not meet PDB standard?
-
Answer
In both cases, the cost is ZERO. NovoPro is first company in the world which offer a risk-free crystallography service. You only pay for success, nothing else.
- 3. What should I do to prepare for the crystal screen?
-
Answer
Before initiating a crystal screen, it is useful to establish “standard” soaking conditions to understand the tolerability of your crystal to ligands. Because this library is provided as a dry film, one can eliminate the need to test for co-solvent tolerance by not adding organic co-solvent. If your crystallization conditions permit, one may test soaking in cryo-solution to remove the added step of cryoprotection before freezing. In principle, ligands should diffuse into crystals very rapidly (minutes) but it can take longer to reach equilibrium for low solvent content crystals or partially occluded active sites. NovoPro typically soaks overnight, but this is a parameter that should be optimized for each crystal system.
- 4. My crystals cracked when I added them to the soak, what should I do?
-
Answer
Crystal cracking can result from a few different scenarios. Typically, one wants to reduce the strain on the crystal by soaking at lower compound concentration for shorter time periods. Crystals may also be uniquely sensitive to certain compounds or compound classes. These compounds may bind at crystal contacts. In this case, gentler soaking conditions may be applied to these specific compounds.
If you have not tested soaking for a ligand known to bind to your binding site, the best path forward is to complete this experiment, even if it is a fragment of the ligand or substrate. This will suggest if ligand binding at the active site results in a conformational shift that results in crystal cracking.
In some cases, a ligand binding at previously unknown secondary binding sites can result in crystal cracking. If a subset of ligands results in crystal cracking and a co-crystal structure cannot be obtained by soaking, one may set-up more traditional co-crystallization experiments with the ligand to identify a new crystal packing. Ligand may be diluted out of the soaking solution or dry compound may be purchased from NovoPro for additional testing.
If a subset of mixtures results in crystal cracking, one may obtain individual compounds to deconvolute and identify the ligand resulting in the crystal crack.
- 5. Some of the compounds are not soluble in my mother liquor. What should I do?
-
Answer
The crystal soak experiment establishes an equilibrium between free ligand and bound ligand. If some of the ligand is not fully dissolved, then the equilibrium includes undissolved ligand, dissolved ligand, and bound ligand. It has been demonstrated that this is not an issue in crystal screening. To ensure that your soak reaches full equilibrium including bound ligand, we suggest soaking these compounds overnight (if the crystal tolerates it).
- 6. How can I use your plates to test different soaking conditions?
-
Answer
We suggest preparing the 20mM stock as described and completing serial dilutions of compound into a separate plate to test different soaking concentrations.
- 7. What if I cannot obtain crystals in the absence of ligand? Can I still do a crystal screen?
-
Answer
Yes! In many cases, co-crystals may be obtained in the presence of a weakly binding ligand. One may soak this ligand out and soak a new ligand into the crystals. This can work even for very tightly binding ligands, but it may require very long soak times for the co-crystal in mother liquor containing no ligand.
Alternatively, Zenobia’s plates may also be used for co-crystallization experiments by adding protein and mother liquor directly to the drop and incubating as a typical sitting drop experiment. Compound concentration may be varied by serial dilution as described above. (back to top)
- 8. I didn't get any hits? How is this possible?
-
Answer
If this is a known druggable target, this issue may be technical. If you have not verified that a known ligand binds to your site, complete this experiment. If you do not have a known ligand:
First, closely examine all of your electron density maps for a feature that is consistent but larger than a water molecule. It is possible that a cryo-molecule or crystallization reagent is binding to the ligand binding site and blocking binding of other ligands.
Look closely at the crystal packing, is it possible that the active site is blocked by a neighboring molecule?
If this is a “difficult” target, do you have a binding pocket that can accommodate a ligand? Is your target undruggable?
To verify a lack of hits, one may also set-up co-crystallization experiments.
- 9. How pure does my sample have to be? For both biophysical characterization and crystallography experiments, the highest level of purity obtainable for your macromolecule is crucial to obtaining high quality data. How pure is pure enough?
-
Answer
This is difficult to answer, but if you can provide material that is 95% or higher purity as estimated by assays such as gel electrophoresis, mass spec. analysis or other analytical chromatography technique, this will give the best chance of success.
- 10. What Buffer Should I Use?
-
Answer
Typical/Example buffers by assay:
Crystallography- 10 mM Tris pH 8.0, 25-100 mM NaCl
SPR- 50 mM Hepes pH 7.4, 150 mM NaCl, 1 mM EDTA, 0.05% Tween/ or PBST (avoid components with high refractive indices and high viscocities when possible such as DMSO, sucrose, glycerol etc.)
CD spectroscopy- low concentrations of UV-Vis inactive buffering agents. Avoid chloride, citrates, MOPS, immidazole and DTT as these components absorb in the visible and/or UV region. Phosphate and Tris buffers are common (do not pH with HCl). De-gas all buffers prior to analysis.
AUC- PBS is commonly used. Ensure that all biomacromolecules are dialyzed into the same batch of buffer. Provide the Core staff with a small volume of your dialysis buffer for dilutions and reference channels.
ITC –PBS or Tris are commonly used depending on the type of interaction that you are studying (proton dependent or independent). Dialyze all samples into the same batch of buffer and provide a sample of that buffer to the core staff along with your sample. Degas all buffers and samples immediately prior to analysis.
KinExA- PBS is typically used as a buffer. BSA will be added to samples prior to analysis.
DSF- flexible buffers, but HEPES is typical.
- 11. How much sample do I need to provide for structural experiments?
-
Answer
Very generally speaking, 10-20 mg for crystallography, 1-10 mg for ITS/DSF/CD/KinExA and 100 ug-1mg for SPR.
- 12. How should I deliver my macromolecule?
-
Answer
If you are delivering your protein to us personally, please bring it on ice if it is in a thawed state already or bring it frozen on dry ice if it was previously frozen or a lyophilized powder. If you are shipping us your material, email us in advance to ensure that we have someone here to receive it. Ship it overnight either on ice or frozen on dry ice.